Tools
Software tools developed by CEIRR investigators for researching influenza and related pathogens.
-
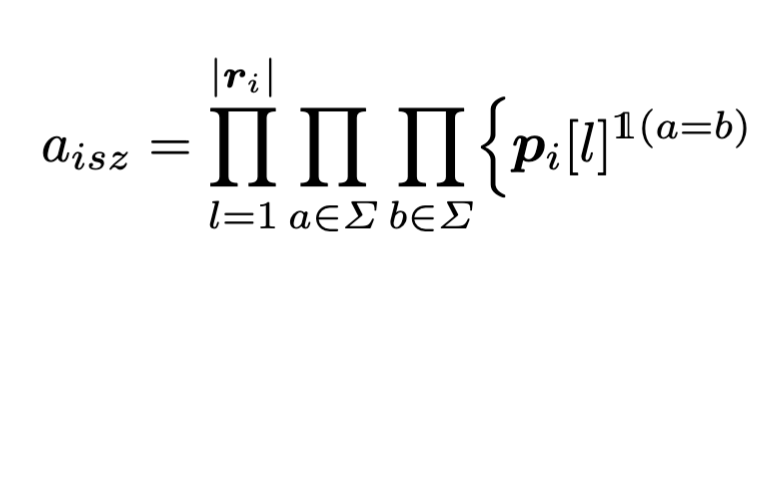
PREMISE
PREMISE is a a probabilistic framework for source assignment of viral Illumina reads.
-

SARS-CoV-2 Variant Nowcast Hub
Weekly reports summarize variant predictions across US states
-
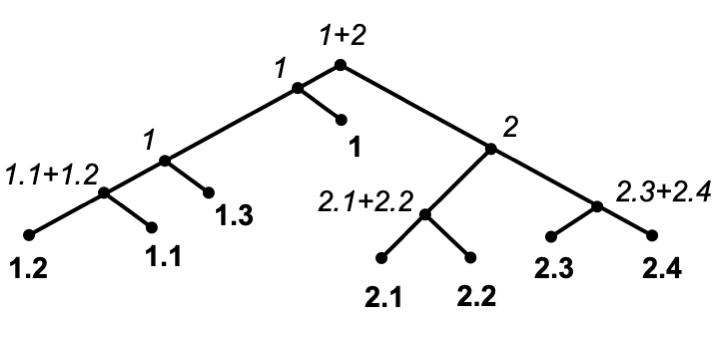
Splicer
Splicer conducts phylogenetic placement on datasets with millions of reference sequences in sub-linear time and can automatically classify new sequences via pre-defined classification file
-
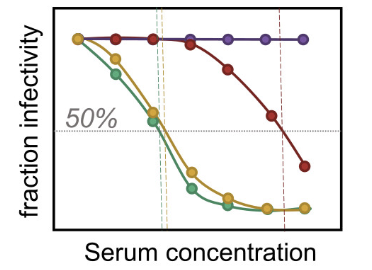
seqneut-pipeline 6.3.0
Pipeline for analyzing high-throughput sequencing-based neutralization assays
-
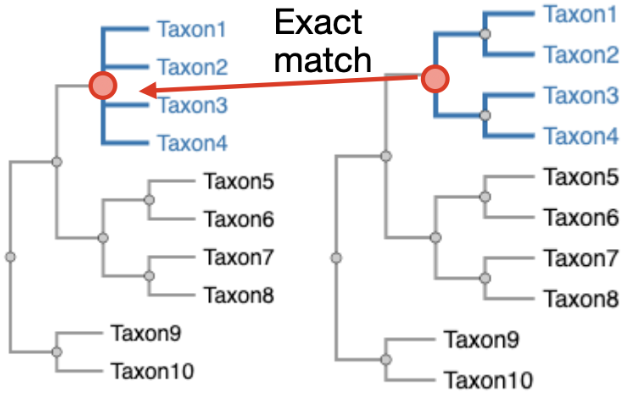
TreeSort Cladeset Mapping
Map TreeSort annotations back onto Nextstrain tree
-

Neutralization Titers in Nextstrain
Bloom lab’s “near real-time” neutralization titers from human sera projected onto Nextstrain trees, alongside raw neutralization measurements
-

Influenza Clade Forecasts
Multinomial logistic regression based variant frequency forecasts for all three seasonal influenza subtypes
-

Phylo-rs
Fast, extensible, general-purpose library for phylogenetic analysis and inference written in the Rust programming language. Phylo-rs provides a robust set of memory-efficient data structures and elementary phylogenetic algorithms
-

immunaut
An innovative predictive platform harnessing unsupervised AI to reveal hidden patterns and forecast outcomes from baseline data
-

PANDORA
Software suite unleashing the power of machine learning for deep insights in omics data, transforming biomedical research possibilities
-
octoFLU
Script that labels phylogenetic clades based on the clade of the nearest neighbor using patristic distances determined from the tree
-
Risk Assessment
A collection of tools curated by the Phylodynamics and computational modeling subgroup for the CEIRR Risk Assessment Pipeline
-

SARS-CoV-2 Forecasts
Forecasts of variant frequencies based on growth advantages estimated through multinomial logistic regression
-

TreeSort
Reassortment inference
-

Analysis of the H5 Outbreak in Cattle
Preliminary report on genomic epidemiology of the 2024 H5N1 influenza A virus outbreak in U.S. cattle
-

H5 HA DMS mutations
Visualization of effects of mutations to H5 hemagglutinin (HA)
-

DMS-viz
Platform for structure-based visualizations of deep mutation scanning data
-

H5Nx in North America Nextstrain Narrative
High path avian influena outbreak dynamics in North America: 2021-present
-

SARS-CoV-2 Mutation Fitness
Estimates of the fitness effects of mutations to all SARS-CoV-2 proteins
-

SARS-CoV-2 Antibody Escape Map
An interactive platform to visualize large amounts of deep mutational scanning data on antibody escape
-

H5Nx Nextclade Datasets
Clade assignment and quality control for H5Nx, H5Nx clade 2.3.4.4, and H5Nx clade 2.3.2.1
-
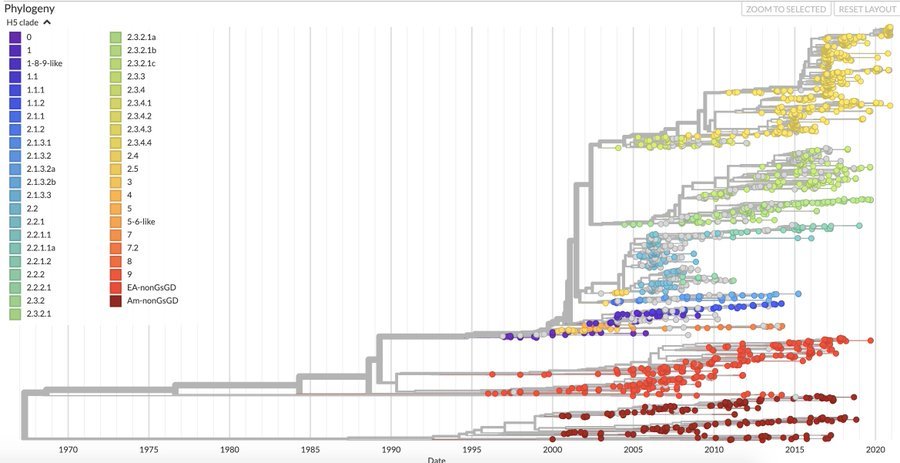
H5Nx Nextstrain Builds
Avian influenza sequences in builds are annotated and colored by whether sequences were sampled from domestic or wild birds, the avian order of the host species (e.g., Anseriformes, Galliformes, etc…), and North American flyway.
-

Imprinting Calculator
imprinting is an R package to reconstruct birth year-specific probabilities of imprinting to each subtype of influenza A, H1N1, H2N2, H3N2. Reconstructions are done following the methods of (Gostic et al. 2016).
-

PARNAS
PARNAS identifies taxa that best represent diversity on a phylogenetic tree and can be used to 1) Select most representative taxa, 2) Downsample a large phylogeny while optimally preserving the underlying diversity, 3) Reduce redundancy among genetic/genomic sequences, 4) Identify key diversity groups on a phylogeny.
-
classLog
Implementation of logistic regression for classification of sequences based on a reference set. Classlog is designed to train logistic regression classifiers based on genetic information. Trained classifiers can then be used to assign classification future clades with linear time complexity.
-

Nextstrain Groups
Nextstrain Groups is a feature that allows research labs, public health entities, and other organizations to share their Nextstrain datasets and narratives directly on nextstrain.org
-

Nextstrain Seasonal Influenza Workflow
Nextstrain workflow for seasonal influenza analysis
-

Nextstrain
Nextstrain is an open-source project to harness the scientific and public health potential of pathogen genome data.
-

polyclonal
polyclonal models mutational escape from polyclonal antibodies using deep mutational scanning data.
-

Flu Strain Compare
Flu Strain Compare generates visualizations of mutations between pairs of HA sequences. Given two sequences as input, it will output a figure or animation highlighting amino acid and PNGS changes on a representative HA crystal structure. H1pdm and H3 strains are supported.
-
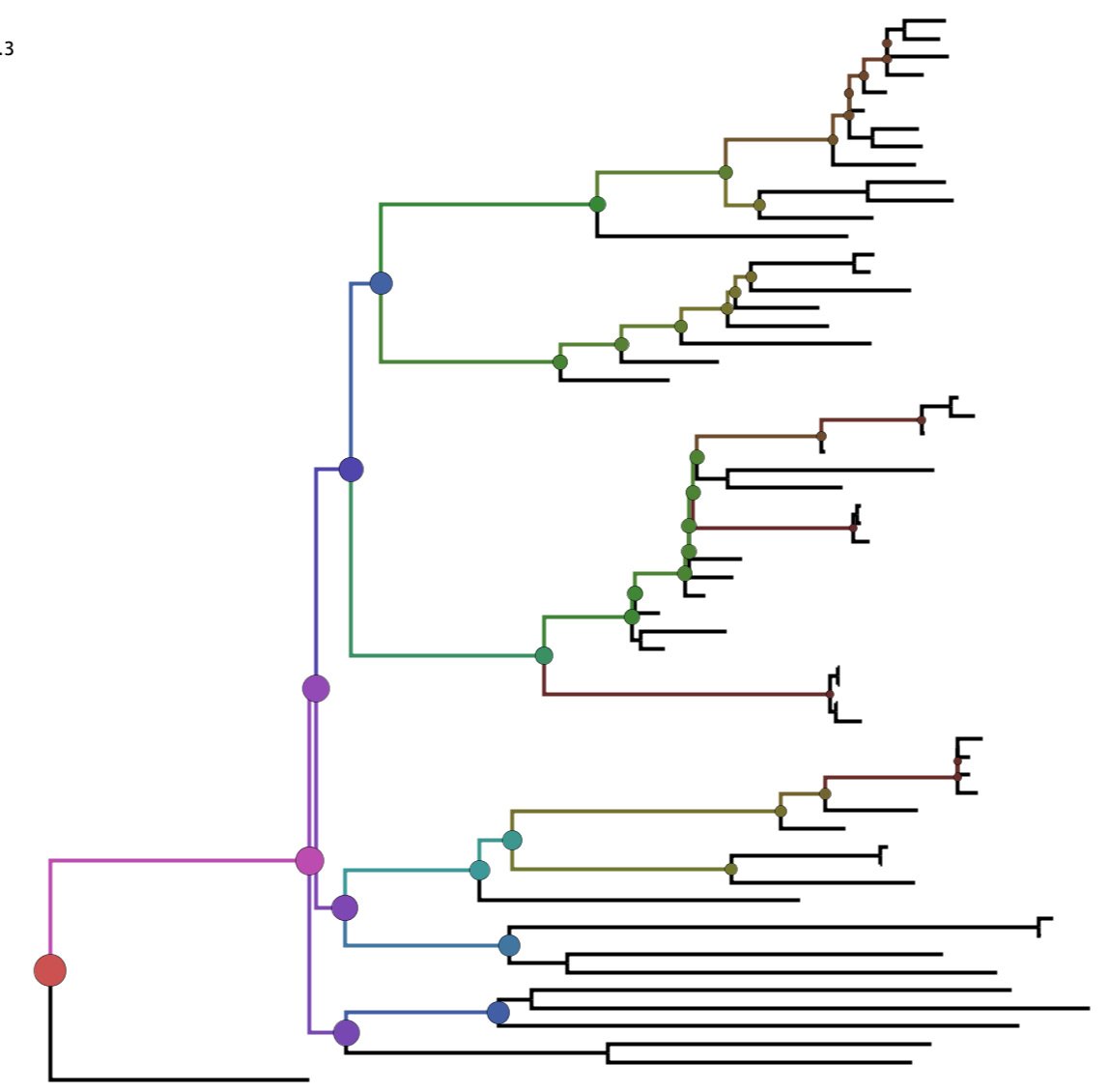
PD-stats
PD-stats quantifies phylogenetic diversity and associated descriptive statistics for all clades in a phylogeny. The tool can be used to identify clades of viruses that have high relative genetic diversity, or groups of viruses that are more diverse than expected by chance.
-

smot
smot is a command-line application for manipulating phylogenies that includes algorithms for subsampling, filtering and classifying phylogenetic trees.
-

Flu Mutation Explorer
The Flu Mutation Explorer provides influenza A virus phylogenies and associated metadata, plus a database of mammalian adaptation amino acid replacements. Phylogenies are created by clustering Genbank sequences for the 8 genome segments and selecting a representative sequence per cluster, which are then used to construct phylogenetic trees.